Iria Pena González.
Estudiante 6º medicina. Universidade de Santiago de Compostela
Emiliano Fdez-Obanza Windscheid.
Cardiólogo. Unidad de Arritmias. Hospital Álvaro Cunqueiro. Vigo
Sergio Raposeiras Roubín.
Cardiólogo clínico. Profesor asociado USC. Hospital Alvaro Cunqueiro. Vigo
Tema 2: Dislipemias
INTRODUCCIÓN
Las dislipemias constituyen uno de los principales factores de riesgo cardiovascular modificables. El colesterol unido a lipoproteínas de baja densidad (LDL) es el principal responsable del depósito lipídico en la pared arterial, iniciando y perpetuando el proceso aterosclerótico. El manejo adecuado de las dislipemias, junto con el control de otros factores de riesgo, ha demostrado reducir de forma muy significativa la incidencia de infarto de miocardio, ictus y muerte cardiovascular.
En este tema abordaremos el metabolismo del colesterol, la fisiopatología de la aterosclerosis, los fármacos hipolipemiantes disponibles, las estrategias de tratamiento según las guías europeas (ESC/EAS 2019, con actualizaciones posteriores) y los objetivos de cLDL según la categoría de riesgo.
DISLIPEMIAS
INTRODUCCIÓN AL METABOLISMO DEL COLESTEROL
El colesterol es un lípido esencial para la estructura de membranas y la síntesis de hormonas, pero su exceso en sangre, especialmente unido a lipoproteínas de baja densidad (LDL), es un factor de riesgo aterogénico clave.
Transporte de colesterol:
- VLDL/IDL/LDL: El hígado produce VLDL, que se transforman en LDL. Estas partículas transportan colesterol a los tejidos periféricos. La apoproteína B100 (ApoB100) es la proteína estructural de las LDL y se une al receptor de LDL (LDL-R) en la membrana celular.
- Receptor de LDL (LDL-R): Es la principal vía de eliminación del colesterol LDL de la sangre. Tras la unión LDL-LDL-R, el complejo se internaliza por endocitosis. El colesterol se libera en la célula y el receptor puede reciclarse a la membrana. Cuantos más receptores funcionales, menor es el LDL circulante.
- HDL (High-Density Lipoprotein): Realiza el transporte inverso de colesterol, capturando el colesterol sobrante de los tejidos y devolviéndolo al hígado para su excreción (vía biliar). Por ello, se le conoce como “colesterol bueno”
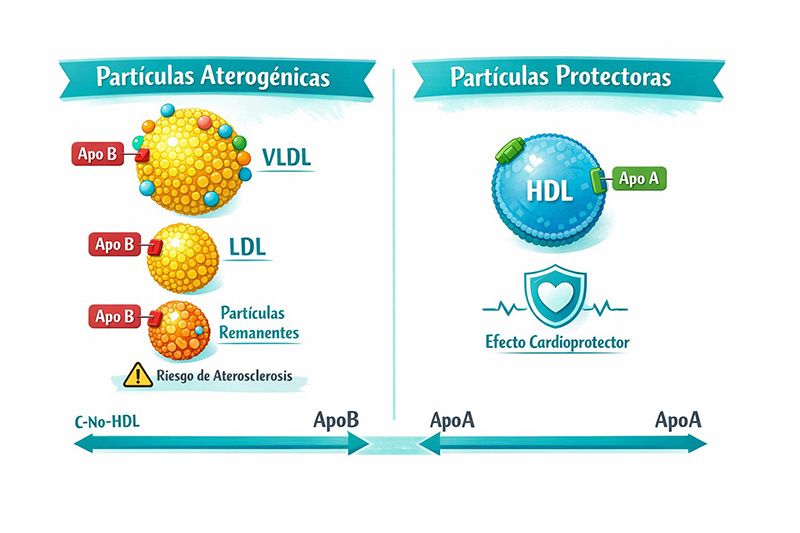
Hipercolesterolemia Familiar (HF)
Es una enfermedad genética autosómica dominante, generalmente debida a mutaciones en el gen del receptor de LDL (LDL-R), lo que reduce su número o funcionalidad. Esto provoca un aumento muy significativo del colesterol LDL circulante desde el nacimiento, llevando a una aterosclerosis acelerada y enfermedad cardiovascular prematura.
- Cribado: Se debe realizar screening de hipercolesterolemia familiar a todo individuo que presente un LDL > 190 mg/dL (especialmente si es < 45 años en hombres o < 55 años en mujeres, o con antecedentes familiares de enfermedad coronaria prematura). En niños el rango sería >160mg/dl.
DESARROLLO DE LA ATEROSCLEROSIS
La aterosclerosis es una enfermedad inflamatoria crónica de la pared de las arterias de mediano y gran calibre. Comienza en la infancia y progresa silenciosamente durante décadas.
Factores influenciadores:
- Genéticos: Antecedentes familiares de cardiopatía isquémica prematura.
- Adquiridos (modificables): Dislipemia, hipertensión arterial, diabetes, tabaquismo, etc.
Estadios evolutivos:
Disfunción endotelial → Estría grasa → Placa de ateroma → Complicación clínica
Disfunción endotelial:
Inducida por factores de riesgo (tabaco, hipertensión, hipercolesterolemia, diabetes). Aumenta la permeabilidad a las LDL, que se depositan en la íntima.
Estría Grasa (Lesión inicial):
En presencia de disfunción endotelial (inducida por factores de riesgo cardiovascular), aumenta la permeabilidad de la íntima. Las partículas de LDL entran y se acumulan en la capa íntima, donde son modificadas (oxidación). Este proceso es inicialmente reversible y asintomático.
Placa de Ateroma (Lesión establecida):
Cuando el estímulo aterogénico persiste, se produce una respuesta inflamatoria:
- Acúmulo lipídico y formación de células espumosas: Los macrófagos fagocitan las LDL oxidadas, convirtiéndose en células espumosas, que son el sello de la lesión temprana.
- Inflamación y necrosis: La liberación de citoquinas por los macrófagos perpetúa la inflamación. La muerte de estas células forma un núcleo lipídico necrótico.
- Fibrosis: Las células musculares lisas migran desde la media a la íntima, proliferan y sintetizan colágeno, formando una cápsula fibrosa que rodea el núcleo lipídico.
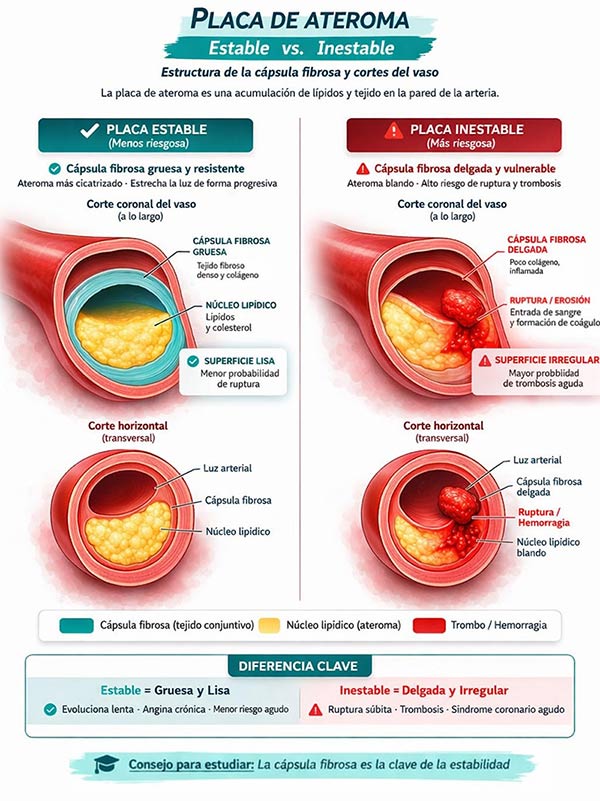
Tipos de placa según su morfología y estabilidad:
| CARACTERÍSTICAS | Placa Estable | Placa Vulnerable / Inestable |
| CÁPSULA FIBROSA | Gruesa, rica en colágeno y células musculares lisas. | Delgada, pobre en células musculares lisas, infiltrada por macrófagos |
| NÚCLEO LIPÍDICO | Pequeño | Grande |
| PROBABILIDAD DE ROTURA | Baja | Alta. Es la responsable de la mayoría de SCA. |
| ESTENOSIS | Puede causar estenosis | A menudo no es estenótica (<50%), pero es más peligrosa por su tolerancia a la rotura |
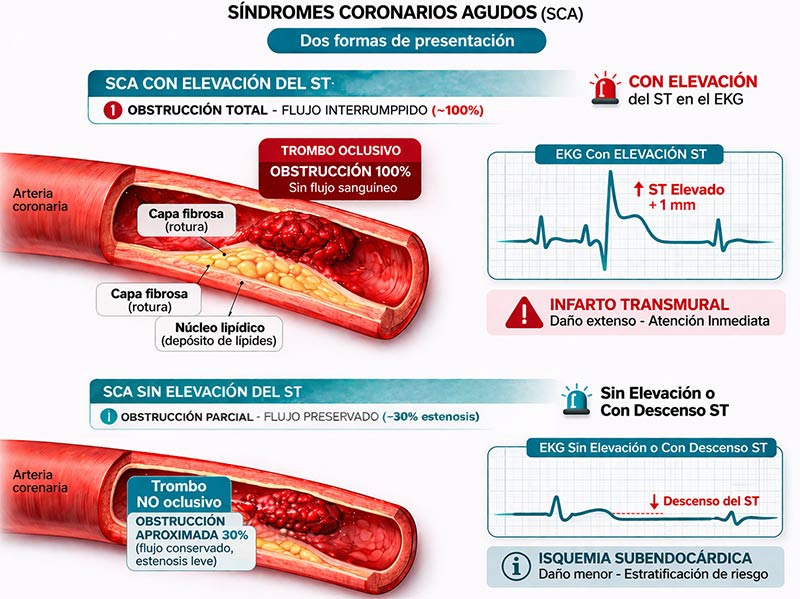
DE LA PLACA A LA CLÍNICA: SÍNDROMES CORONARIOS AGUDOS (SCA)
La rotura o erosión de una placa vulnerable expone su contenido trombogénico al torrente sanguíneo, desencadenando la hemostasia:
- Activación plaquetaria: Las plaquetas entran en contacto con el factor tisular y el colágeno subendotelial. Se activan y expresan la glucoproteína IIb/IIIa en su membrana, que se une al fibrinógeno formando puentes entre plaquetas (agregación).
- Cascada de coagulación: Se activa, culminando en la formación de trombina, que convierte el fibrinógeno en fibrina, estabilizando el trombo.
El grado de obstrucción del flujo sanguíneo determinará el tipo de SCA:
- SCA con elevación del ST (SCACEST): Corresponde a una oclusión completa y persistente del vaso por un trombo rico en fibrina. Esto provoca una isquemia transmural que lleva a necrosis miocárdica (infarto). Puede presentarse como infarto o como muerte súbita (frecuentemente por fibrilación ventricular).
- SCA sin elevación del ST (SCASEST): Hay un trombo que ocluye de forma intermitente o incompleta (trombo lábil). No hay oclusión total sostenida, por lo que no hay elevación del ST. La isquemia es subendocárdica. Es más común en pacientes con placas estenóticas previas que tenían angina estable.
DIAGNÓSTICO POR IMAGEN DE LA PLACA
- Arteriografía Coronaria (Coronariografía): Es la técnica de referencia para visualizar la luz arterial. Mediante un catéter se inyecta contraste y se obtienen imágenes con rayos X (luminograma). Permite identificar estenosis significativas.
- Importante: Si una coronaria no está críticamente obstruida (estenosis < 50-70%), no se coloca un stent. El tratamiento se basa en cambios de hábitos y terapia médica óptima para estabilizar la placa.
- Técnicas avanzadas: La resonancia magnética, la tomografía de coherencia óptica (OCT) y la ecografía intracoronaria (IVUS) permiten visualizar la pared arterial y caracterizar el tipo de placa (estable vs. vulnerable).
TRATAMIENTO DE LAS DISLIPEMIAS
El objetivo principal es reducir el colesterol LDL (cLDL), ya que es el principal responsable de la aterogénesis.
TRATAMIENTO NO FARMACOLÓGICO
- Dieta mediterránea rica en frutas, verduras, cereales integrales, pescado azul, aceite de oliva virgen extra y frutos secos.
- Ejercicio físico regular (≥150 minutos/semana de actividad aeróbica moderada).
- Abandono del tabaco.
- Control del peso (IMC <25 kg/m²; perímetro de cintura <94 cm en hombres, <80 cm en mujeres).
TRATAMIENTO FARMACOLÓGICO
A. Estatinas (Inhibidores de la HMG-CoA reductasa)
Son el pilar del tratamiento. Reducen la síntesis endógena de colesterol en el hígado, lo que aumenta la expresión de receptores LDL en los hepatocitos y, por tanto, la captación y eliminación de LDL circulante.
- Efectos: Reducción de cLDL del 20-55% (dependiendo de dosis y potencia), aumento moderado de HDL (5-15%), reducción de triglicéridos.
- Alta potencia (reducción cLDL ≥ 50%):
- Rosuvastatina: 10, 20, 40 mg/día.
- Atorvastatina: 40, 80 mg/día.
- Seguridad: Generalmente seguras. Efectos secundarios: mialgias, elevación de transaminasas (leve, suele ser dosis-dependiente), rara vez diabetogenicidad (pequeño aumento del riesgo de diabetes tipo 2, especialmente en pacientes con factores de riesgo).
B. Ezetimiba
Inhibe el transportador Niemann-Pick C1-Like 1 (NPC1L1) en el borde en cepillo del intestino delgado, reduciendo la absorción de colesterol dietético y biliar.
- Efectos: Reduce cLDL en un 15-20%.
- Sinergia: La combinación estatina + ezetimiba tiene un efecto sinérgico, logrando una reducción de cLDL de hasta el 75%, actuando sobre dos vías complementarias: síntesis hepática y absorción intestinal.
C. Inhibidores de PCSK9 (iPCSK9)
Mecanismo: PCSK9 es una proteína que se une al complejo LDL-R y lo dirige hacia la degradación lisosomal, reduciendo el reciclaje del receptor. Al inhibir PCSK9, se aumenta el número de receptores LDL funcionales en la membrana del hepatocito, incrementando drásticamente la captación de LDL.
- Fármacos:
- Anticuerpos monoclonales: Alirocumab, Evolocumab. Administración subcutánea cada 2 semanas o mensual. Reducción de cLDL del 50-60% adicional sobre el tratamiento base.
- ARN de interferencia (siARN): Inclisirán. Inhibe la síntesis hepática de la proteína PCSK9. Administración subcutánea cada 6 meses (dos dosis al año).
- Indicación (coste elevado): Pacientes de muy alto riesgo con enfermedad cardiovascular establecida que, a pesar de recibir estatina de alta intensidad + ezetimiba, no alcanzan los objetivos de cLDL (p. ej., cLDL > 100 mg/dL en prevención secundaria).
D. Ácido Bempedoico
Profármaco que se activa en el hígado (no en el músculo) inhibiendo la ATP-citrato liasa, una enzima upstream de la HMG-CoA reductasa.
- Ventaja: Al no activarse en el músculo, no produce mialgias.
- Efectos: Reduce cLDL un 18-23% (en combinación con estatinas). Indicado en pacientes intolerantes a estatinas o en aquellos de riesgo muy alto que no alcanzan objetivos con estatina + ezetimiba.
E. Fibratos
Activan los receptores PPAR-α, aumentando la lipólisis de triglicéridos (TG) ricos en VLDL.
- Efectos: Potente reducción de TG, ligero aumento de HDL.
- Indicación: Su recomendación es más débil (IIb) en las guías actuales. Se reservan para subgrupos con dislipemia aterogénica (TG elevados, HDL bajo) o hipertrigliceridemia severa (riesgo de pancreatitis).
F. Otros fármacos
- Icosapento etilo (Éster etílico del ácido eicosapentaenoico): Ácido graso omega-3 de alta pureza. A dosis de 2g/12h, en combinación con estatinas, reduce el riesgo de eventos CV en pacientes de alto/ muy alto riesgo con TG elevados (135-499 mg/dL). (Recomendación IIa B).
- Volanesorsén: Oligonucleótido antisentido anti-ApoC-III. Indicado en pacientes con hipertrigliceridemia severa (> 750 mg/dL) por síndrome de quilomicronemia familiar (SQF) para reducir TG y el riesgo de pancreatitis.
- Evinacumab: Anticuerpo monoclonal anti-ANGPTL3. Indicado en pacientes con hipercolesterolemia familiar homocigota (> 5 años) que no alcanzan objetivos.
- Secuestradores de ácidos biliares (resinas): En desuso por baja eficacia y mala tolerabilidad.
| Categoría de Riesgo | Definición | Objetivo de cLDL |
| MUY ALTO | Enfermedad cardiovascular (ECV) clínica establecida (infarto, ictus, revascularización). Diabetes mellitus con ECV o con daño orgánico. Hipercolesterolemia familiar (HF) con ECV o con otros factores de riesgo. ECV prematura. | < 55 mg/dL y reducción ≥ 50% desde el valor basal |
| ALTO | Diabetes mellitus sin ECV ni daño orgánico. HF sin ECV ni otros factores de riesgo. Pacientes con SCORE2/SCORE2-OP ≥ 10% (riesgo a 10 años). | < 70 mg/dL y reducción ≥ 50% desde el valor basal |
| MODERADO | Pacientes con SCORE2/SCORE2-OP entre 5% y < 10%. | < 100 mg/dL |
| BAJO | Pacientes con SCORE2/SCORE2-OP < 5%. | < 116 mg/dL |
*Fuente: Guía ESC/EAS 2019 [1], con escalas SCORE2 [2]. *
ALGORITMO DE TRATAMIENTO
A. Prevención Secundaria y Muy Alto Riesgo
- Inicio: Estatina de alta intensidad (atorvastatina 40-80 mg o rosuvastatina 20-40 mg).
- Evaluación a las 4-6 semanas: Si no se alcanza el objetivo de cLDL (<55 mg/dL), se añade ezetimiba.
- Segunda evaluación: Si persiste sin alcanzar objetivo, se añade un inhibidor de PCSK9.
B. Prevención Primaria
- La decisión se basa en el riesgo estimado (SCORE2/SCORE2-OP) y los niveles basales de cLDL. Se inicia con estatina de intensidad moderada o alta según el riesgo.
C. Situaciones Especiales
- Pacientes con Síndrome Coronario Agudo (SCA): Se recomienda intensificar el tratamiento hipolipemiante durante la hospitalización inicial. En pacientes que no recibían tratamiento, se puede considerar iniciar directamente la combinación de estatina de alta intensidad + ezetimiba durante el ingreso (Recomendación IIa B).
- Enfermedad vascular recurrente: En pacientes que sufren un nuevo evento cardiovascular a pesar de estar en tratamiento óptimo, se pueden considerar objetivos aún más bajos (< 40 mg/dL) con terapia combinada (incluyendo iPCSK9).
PUNTOS CLAVE
- El receptor de LDL es clave: Su función determina los niveles de colesterol circulante. La PCSK9 es su principal regulador negativo.
- No toda placa es igual: La placa vulnerable (de cápsula fina y núcleo lipídico grande) es la que causa los SCA, incluso sin ser muy estenótica.
- El objetivo terapéutico es el cLDL: Cuanto más alto es el riesgo del paciente, más bajo debe ser el objetivo de cLDL (incluso < 55 mg/dL en muy alto riesgo).
- Tratamiento escalonado pero dinámico: Se inicia con estatina de alta intensidad en prevención secundaria, y se añade ezetimiba e iPCSK9 si no se alcanzan objetivos en las evaluaciones periódicas.
- Nuevos fármacos: Conocer las indicaciones de fármacos como el ácido bempedoico (sin mialgias), icosapento etilo (eventos CV en TG altos) e inclisirán (dos dosis al año) es esencial en la práctica clínica actual.
BIBLIOGRAFIA
- Mach F, Baigent C, Catapano AL, et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. 2020;41(1):111-188.
- SCORE2 working group and ESC Cardiovascular Risk Collaboration. SCORE2 risk prediction algorithms: new models to estimate 10‑year risk of cardiovascular disease in Europe. Eur Heart J. 2021;42(25):2439-2454.
- Libby P, Ridker PM, Hansson GK. Inflamación y aterotrombosis. J Am Coll Cardiol. 2009;54(23):2129-2138. (Revisión clásica; se puede consultar ediciones actualizadas en textos de fisiopatología cardiovascular).
- Ference BA, Ginsberg HN, Graham I, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: evidence from genetic, epidemiologic, and clinical studies. Eur Heart J. 2017;38(32):2459-2472.
- Sabatine MS, Giugliano RP, Keech AC, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease – FOURIER trial. N Engl J Med. 2017;376(18):1713-1722.
- Cannon CP, Blazing MA, Giugliano RP, et al. Ezetimibe added to statin therapy after acute coronary syndromes – IMPROVE-IT trial. N Engl J Med. 2015;372(25):2387-2397.
- Nissen SE, Lincoff AM, Brennan D, et al. Bempedoic acid and cardiovascular outcomes in statin‑intolerant patients – CLEAR Outcomes. N Engl J Med. 2023;388(15):1353-1364.
- Bhatt DL, Steg PG, Miller M, et al. Cardiovascular risk reduction with icosapent ethyl for hypertriglyceridemia – REDUCE-IT. N Engl J Med. 2019;380(1):11-22.
- Raal FJ, Rosenson RS, Reeskamp LF, et al. Evinacumab for homozygous familial hypercholesterolemia. N Engl J Med. 2020;383(8):711-720.
- Ray KK, Wright RS, Kallend D, et al. Two phase 3 trials of inclisiran in patients with elevated LDL cholesterol – ORION-9, ORION-10, ORION-11. N Engl J Med. 2020;382(16):1507-1519.